DFT计算分析电荷转移方法有哪些?
密度泛函理论(Density Functional Theory,DFT)是一种广泛应用于材料科学、化学和物理学领域的量子化学计算方法。它通过计算系统的电子密度来描述分子或固体的电子结构,从而预测材料的物理和化学性质。在电荷转移研究中,DFT提供了多种工具和方法,用于分析和理解电荷在不同原子或分子之间的转移过程。本文将详细探讨DFT在电荷转移分析中的应用,包括差分电荷密度、Bader电荷、功函数、HOMO-LUMO能隙、静电势和态密度等方法。
差分电荷密度
差分电荷密度(Differential Charge Density,DCD)是DFT中用于分析电荷转移的一种重要方法。它通过比较两个系统之间的电荷密度差异,揭示电荷转移的方向和程度。 在Fe、Co等金属与基底之间的电荷转移中,由于d电子未填满,电荷在金属与基底间双向流动,体现强轨道耦合。而Cu、Zn因d轨道全充满,仅显示绿色耗散区,说明电子从金属单向转移到gC₃N₄,符合其弱还原性。碱金属(Na、Li、K)因电负性极低,绿色耗散区集中在原子周围,证实电子完全离域到基底,形成离子键特征。 非金属中,B在键合区积累电荷,显示共价键形成;O因高电负性吸引电子,导致自身周围强积累而键合区耗散,呈现极性键;S和P则因中等电负性同时存在积累与耗散,反映混合键合特性。整体上,差分图不仅验证了电负性差异驱动的电荷转移方向,还通过空间分布揭示了键合类型(离子/共价/极性),为理解掺杂原子与gC₃N₄的相互作用机制提供了直观电子尺度证据。Bader电荷
Bader电荷是基于Bader提出的“原子中的原子”(Atoms in Molecules,AIM)理论计算的一种电荷划分方法。该理论通过分析电子密度的拓扑结构(如临界点、梯度路径等),将分子或固体中的电荷密度分布划分为不同的原子区域(Bader体积),并通过积分每个区域内的电子密度来确定原子的净电荷。Bader电荷的计算公式为: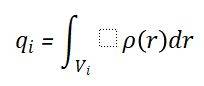 其中,qi表示第i个原子的净电荷,ρ(r)是空间r处的电子密度,Vi是第i个原子的Bader体积。Bader电荷的计算不仅能够确定电荷转移的净量,还能定位电子转移的空间分布,从而全面解析化学键的本质(共价、离子或金属键)。因此,Bader电荷是理论计算和材料模拟中不可或缺的工具,为理解电子结构调控和化学反应机制提供了坚实的理论基础。
其中,qi表示第i个原子的净电荷,ρ(r)是空间r处的电子密度,Vi是第i个原子的Bader体积。Bader电荷的计算不仅能够确定电荷转移的净量,还能定位电子转移的空间分布,从而全面解析化学键的本质(共价、离子或金属键)。因此,Bader电荷是理论计算和材料模拟中不可或缺的工具,为理解电子结构调控和化学反应机制提供了坚实的理论基础。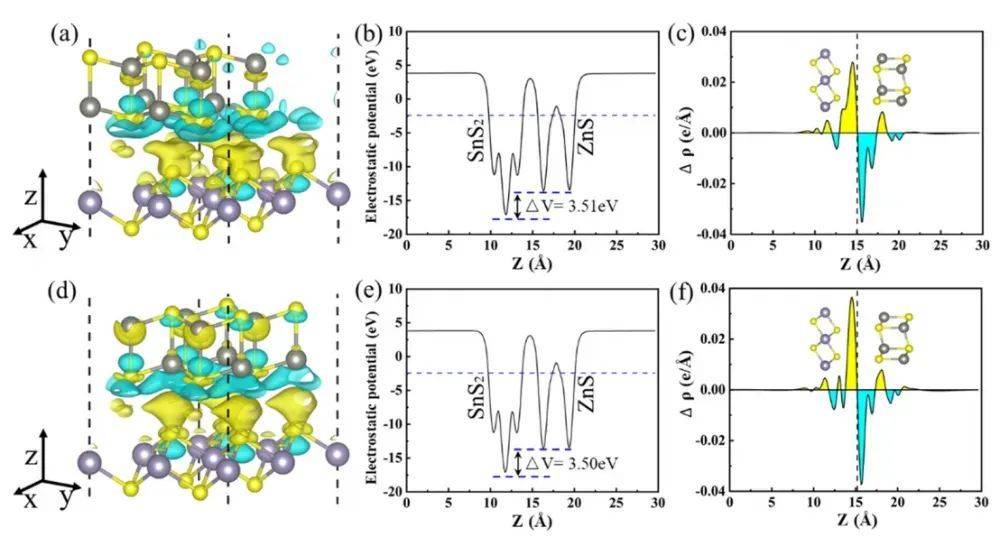
功函数
功函数(Work Function,φ)是指将一个电子从固体内部刚刚移到表面所需克服的最小能量,它等于真空能级与固体费米能级之间的能量差,数学表达式为: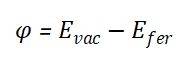 其中,φ表示功函数,Evac是真空能级,Efer是固体的费米能级。真空能级是电子在真空中具有的能量,而费米能级是固体中电子的化学势,反映了电子在固体中的填充水平。当两种不同材料相互接触或发生化学反应时,由于它们的功函数不同,会导致电子在界面处发生转移,以达到新的电子平衡状态。功函数较低的材料会失去电子,功函数较高的材料会获得电子,直到两者的费米能级对齐。通过测量或计算材料的功函数,并分析其在不同条件下(如吸附、掺杂等)的变化,可以推断出材料表面或界面处的电荷转移情况。
其中,φ表示功函数,Evac是真空能级,Efer是固体的费米能级。真空能级是电子在真空中具有的能量,而费米能级是固体中电子的化学势,反映了电子在固体中的填充水平。当两种不同材料相互接触或发生化学反应时,由于它们的功函数不同,会导致电子在界面处发生转移,以达到新的电子平衡状态。功函数较低的材料会失去电子,功函数较高的材料会获得电子,直到两者的费米能级对齐。通过测量或计算材料的功函数,并分析其在不同条件下(如吸附、掺杂等)的变化,可以推断出材料表面或界面处的电荷转移情况。HOMO-LUMO能隙
HOMO-LUMO能隙(HOMO-LUMO Gap)是描述分子得失电子难易程度的重要参数。HOMO(Highest Occupied Molecular Orbital)是分子中最高占据的分子轨道,LUMO(Lowest Unoccupied Molecular Orbital)是分子中最低未占据的分子轨道。HOMO-LUMO能隙的大小反映了分子得失电子的难易程度,能隙越小,分子越容易发生电荷转移。在有机光伏材料中,给体材料的HOMO能级与受体材料的LUMO能级之间的能量匹配对于电荷分离和转移至关重要。
HOMO能级的位置(如Macauromondo的-9.97eV)和LUMO能级(如-0.03eV)的相对能量差决定了电荷转移的方向性——电子通常从高能级的HOMO流向低能级的LUMO。
这种能级匹配关系在远程桥接的2,3,8-核体系中尤为明显,进一步证实了HOMO-LUMO分析在预测电荷转移路径中的重要性。通过HOMO-LUMO能级和能隙的分析,可以清晰地揭示分子内或分子间电荷转移的驱动力和方向性,为理解电子转移过程提供了关键的理论依据。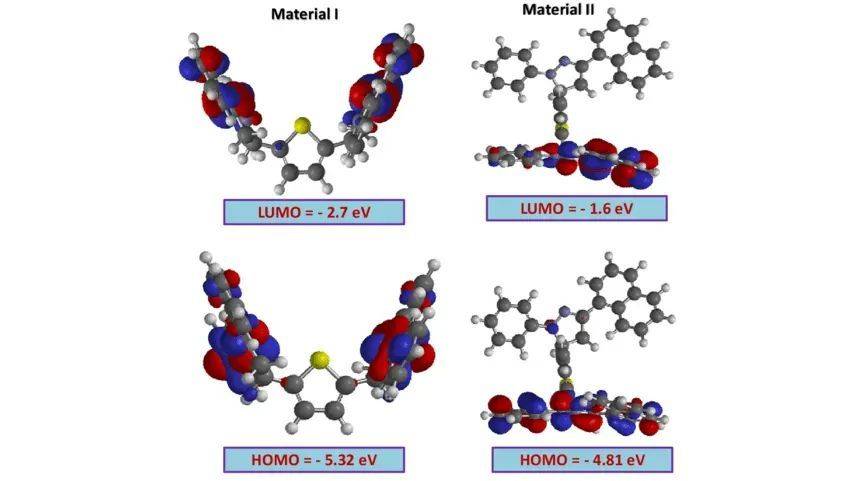
静电势
静电势(Electrostatic Potential,V(r))是指在空间某一点处,将一个单位正电荷从无穷远处移动到该点时所做的功,它是描述体系电子结构的一个重要物理量。在DFT计算中,静电势可以通过求解泊松方程从电子密度计算得到,其数学表达式为: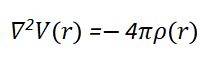 其中,V(r)表示空间r处的静电势,ρ(r)是空间r处的电子密度。静电势反映了体系中电子和原子核的电荷分布对空间电场的影响。当两个组分(如分子与基底)接近时,电荷倾向于从高静电势区(电子供体)流向低静电势区(电子受体)。因此,静电势能够分析电荷转移,关键在于其直观体现电子给/受能力。
其中,V(r)表示空间r处的静电势,ρ(r)是空间r处的电子密度。静电势反映了体系中电子和原子核的电荷分布对空间电场的影响。当两个组分(如分子与基底)接近时,电荷倾向于从高静电势区(电子供体)流向低静电势区(电子受体)。因此,静电势能够分析电荷转移,关键在于其直观体现电子给/受能力。态密度
态密度(Density of States,DOS)是描述材料中电子能级分布的函数,表示单位能量区间内允许的电子量子态数量。态密度可分为总态密度(TDOS)和分波态密度(PDOS),后者进一步分解为原子轨道(s、p、d、f)的贡献,从而揭示不同轨道的电子结构特征。通过投影态密度(PDOS)分析揭示了HF分子在Li₂MnO₃(001)表面吸附时的电荷转移机制。 PDOS结果显示,Mn的d轨道在费米能级附近的贡献几乎未受HF吸附影响,表明Mn未直接参与电荷交换,与Bader电荷分析中Mn仅+0.02e的微小变化一致。关键变化集中在F和H的轨道上:
PDOS结果显示,Mn的d轨道在费米能级附近的贡献几乎未受HF吸附影响,表明Mn未直接参与电荷交换,与Bader电荷分析中Mn仅+0.02e的微小变化一致。关键变化集中在F和H的轨道上:F原子在-7.5eV附近出现新峰,对应其p轨道与表面Li的杂化,证实了F-Li的离子键相互作用;而H原子在-7eV附近的峰则指向H-O键的形成,解释了实验中H从HF解离后与表面O的键合。值得注意的是,随着HF覆盖度增加,F相关峰强度增强(0.06eV处),表明更高覆盖度下F的电子局域化更显著,可能增强表面钝化效应。
此外,费米能级附近未出现金属性新态,说明HF吸附未引入额外导电通道,而是通过F和H的轨道重构实现电荷再分配,降低了材料反应活性。态密度分析因此清晰地揭示了
电荷转移的原子尺度路径:电子从Li流向F形成极性键,同时H与O键合,而Mn保持惰性,这一机制为理解表界面修饰对材料电子结构的调控提供了重要依据。
总结
综上所述,差分电荷密度、Bader电荷等电荷布局、功函数、HOMO-LUMO、静电势和态密度这六种方法从不同角度为DFT计算中分析电荷转移提供了有力的工具。它们各自具有独特的定义和分析原理,在材料科学、化学、物理学等领域有着广泛的应用。通过综合运用这些方法,可以更全面、深入地理解电荷转移过程,为新材料的设计和性能优化提供坚实的理论基础。
在未来的研究中,随着DFT计算方法的不断发展和完善,这些分析方法也将不断拓展其应用范围和深度,为科学研究和技术创新提供更强大的支持。