VASP电荷密度计算流程
VASP自洽计算原理
VASP(Vienna Ab initio Simulation Package)是一款基于密度泛函理论(DFT)的量子力学模拟软件,广泛应用于材料科学、化学、物理学等领域。
其核心计算方法之一是自洽场(Self-Consistent Field, SCF)算法,解决Kohn-Sham方程,该算法通过迭代计算电子波函数和密度,直到系统达到自洽状态。
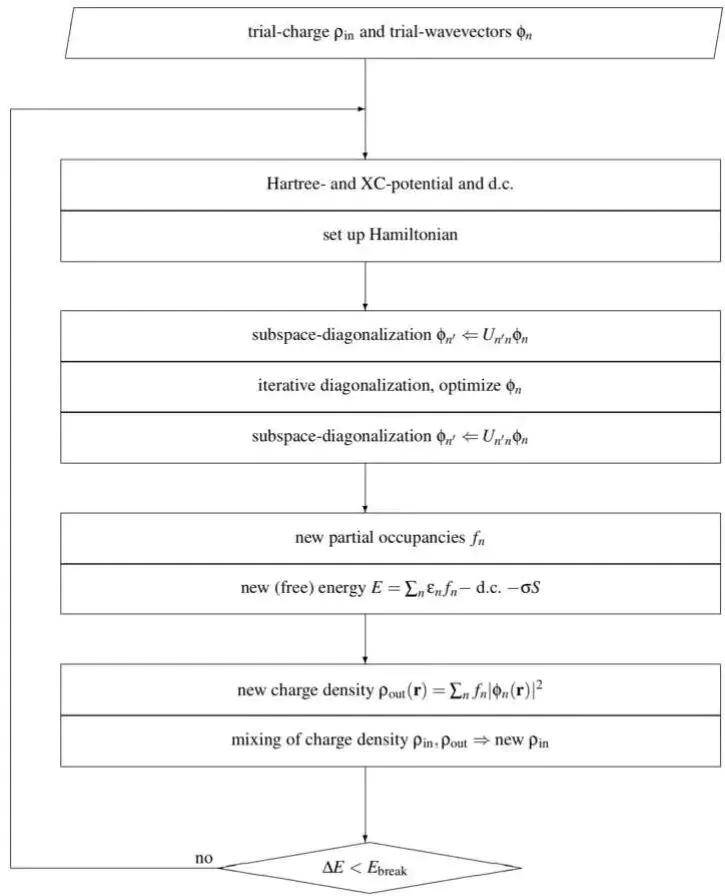
自洽计算是VASP计算中最基础也是最重要的步骤,其目的是找到电子波函数和相应的总能量,使得电子在晶胞中的分布达到自洽。
自洽计算通常包括电荷密度的初始化、波函数的迭代优化以及能量和力的计算。
准备工作
VASP计算需要准备超算连接软件EASYCONNECT与SSH,建模软件VESTA,超算连接软件Winscp。
VESTA软件下载链接
jp-minerals.org/vesta/en/download.html
EASYCONNCT软件下载链接
EasyConnect下载-EasyConnect最新版下载V7.6.7.0
Winscp软件下载链接
Downloading WinSCP-6.5.3-Setup.exe :: WinSCP
VASP输入参数说明
INCAR文件:
ISTART=0 #开始新的任务,随机产生初始波函数
ICHARG=2 #开始新的任务,从原子电荷密度产生体系初始电荷密度
PREC=M #计算精度,决定ENCUT
ISPIN=1 #关闭自旋极化
ALGO=N #确定电子优化的算法
NELM=60 #电子波函数最多计算60步
EDIFF=1E-5 #相邻两步电子迭代的能量差收敛标准
ENCUT=400 #平面波截断能400 eV
IVDW=11 #考虑范德华力修正
IBRION=2 #共轭梯度算法,适用于结构优化
NSW=100 #离子弛豫的步数
ISIF=3 #控制晶格变化,晶格常数优化
EDIFFG=-0.1 #离子弛豫的force的收敛标准
ISMEAR=0 #费米能级附近电子占据数为高斯分布,适合金属、半导体、绝缘体
SIGMA=0.1 #高斯分布展宽0.1 eV
LORBIT = 10 #局域态密度
KPOINTS文件:
Automatic generation #注释行
0 #自动产生K点网格
G #布里渊区K点网格以Gamma点为中心
5 5 5 #K点网格密度
0 0 0 #K点网格中心平移矢量
MgO电荷密度计算
第一步,复制结构优化的CONTCA R,KPOINTS,POTCAR文件
进到之前结构优化的位置,确认一下结构优化已经完成
cd crystal/mgo/opt

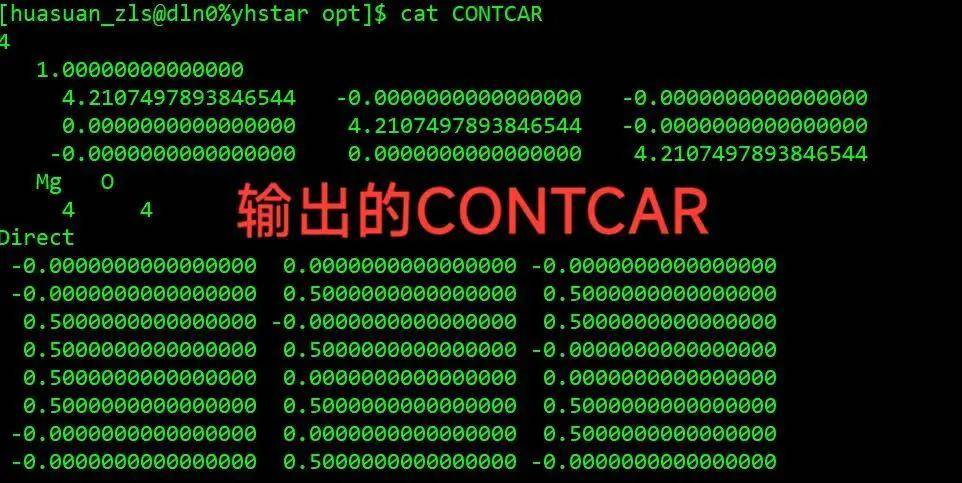
将结构优化的文件夹变成自洽计算的文件夹
把它命名成s,进到 s 里面把 CONTCAR 复制成POSCAR
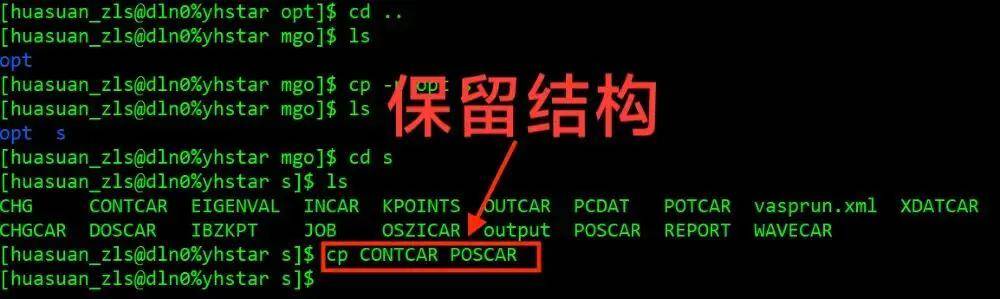
cd ..
cp -r opt s
cd s
cp CONTCAR POSCAR
第二步,修改结构优化的 INCAR 文件
修改 INCAR 文件,让 VASP 执行自洽计算
vi INCAR

第三步,提交自洽计算
sbatch JOB

使用 squeue 命令来看任务队列,已清空。

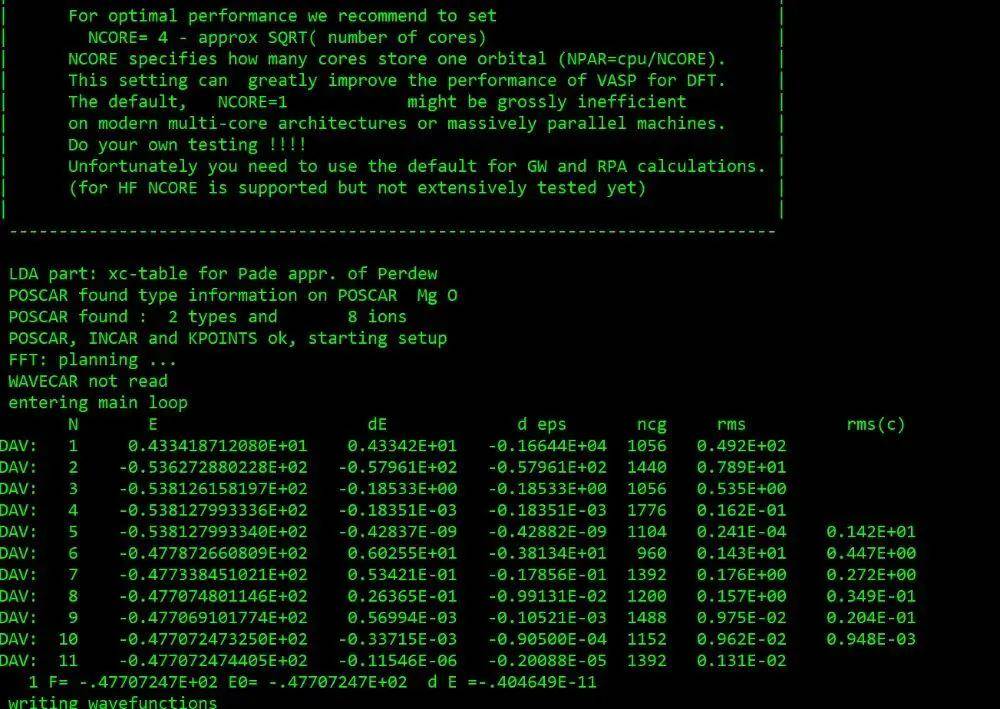
计算完成后查看输出文件output
cat output
第四步,用VESTA读取输出文件CHGCAR并绘制电荷密度图
算完之后下载输出的CHGCAR文件。进到刚才自洽计算的文件夹 crystal/mgo/s,下载CHGCAR
用 VESTA 打开CHGCAR
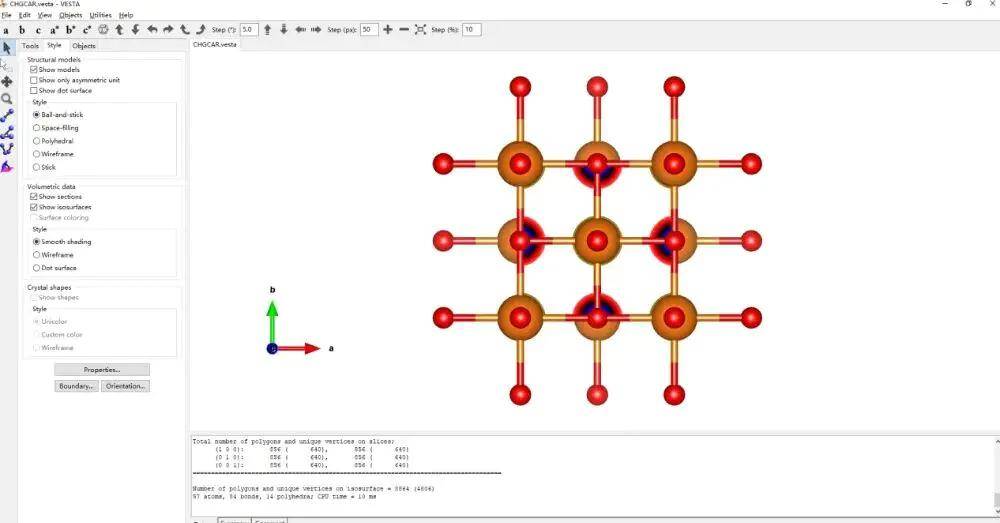
给结构图做一个简单的修饰,加上化学键。
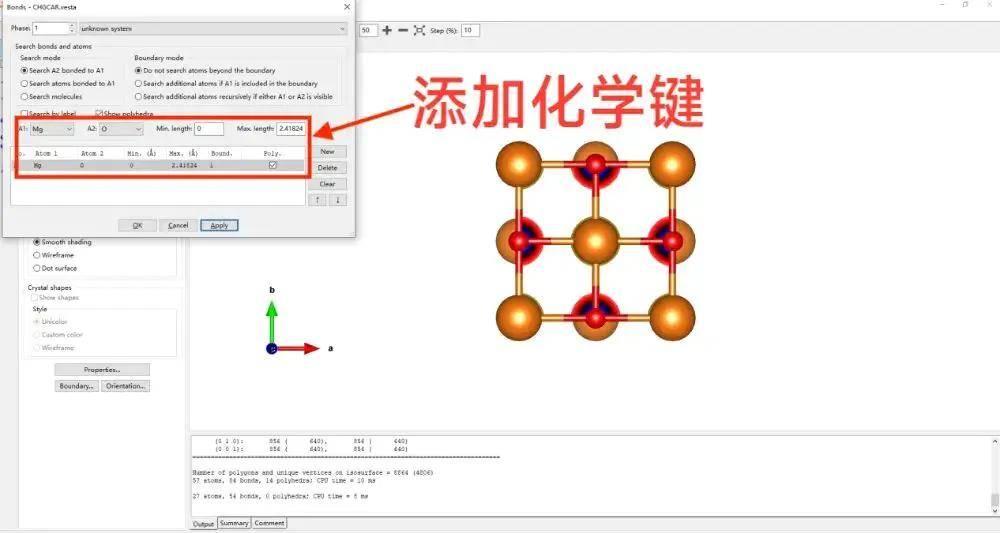
修改电荷,在isosurfaces面板中有一个等值面
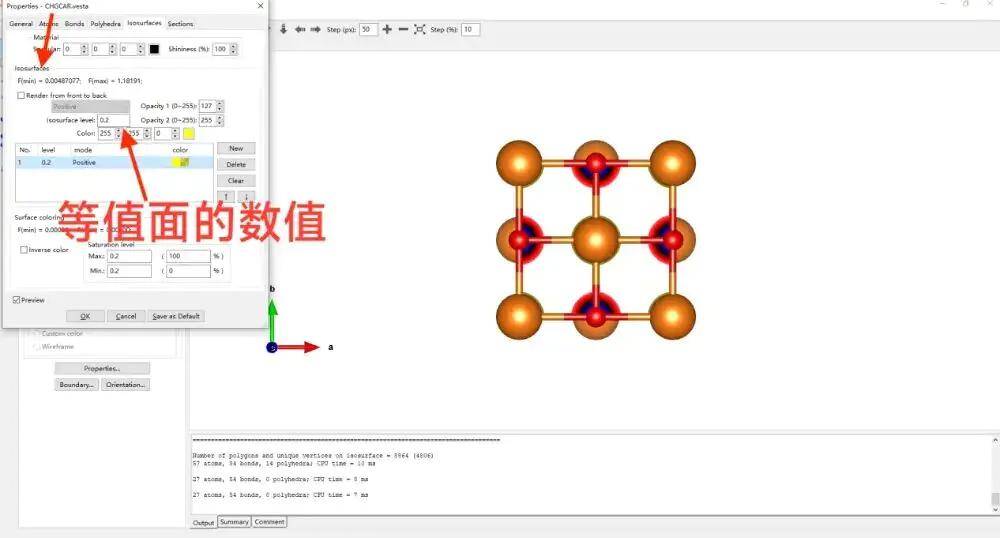
空间当中电荷密度大于该数值的,会显示这种电荷云
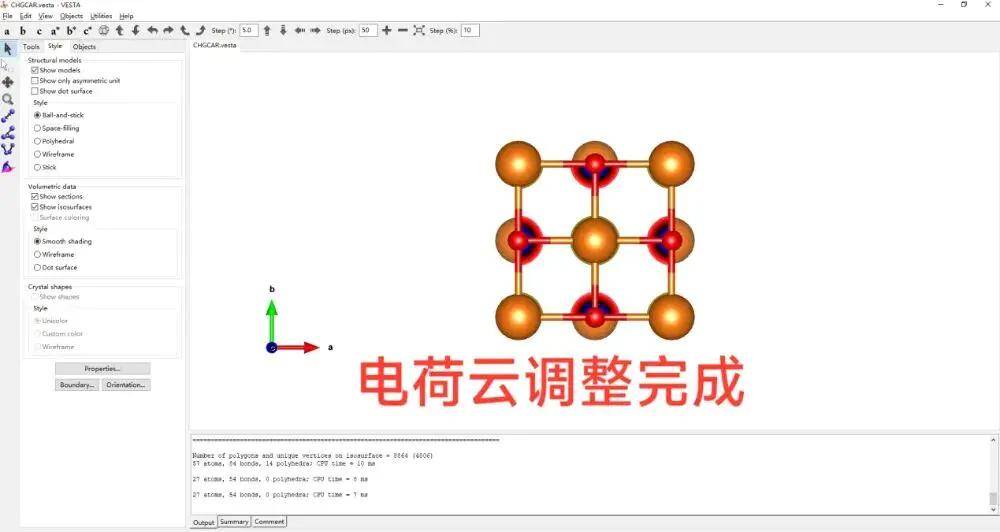
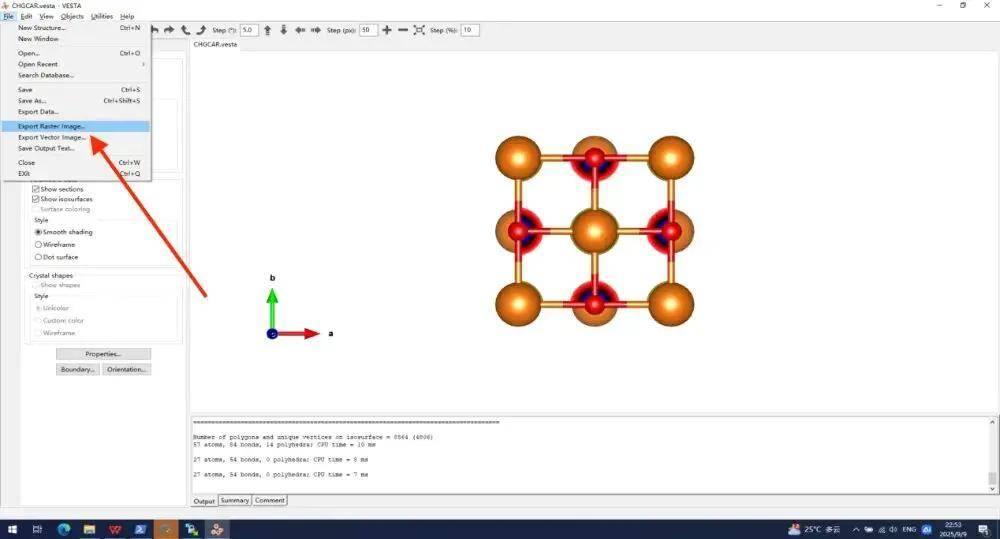
把它叫做CHAGCAR.png 保存。