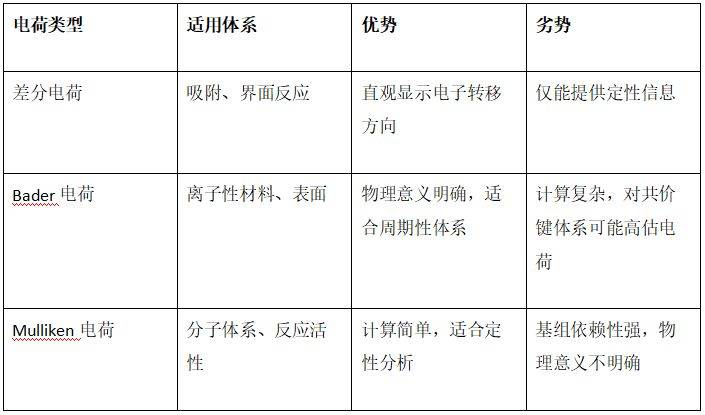
什么是差分电荷、Bader电荷、Mulliken电荷?
在化学和材料科学中,电荷分析是理解分子间相互作用、化学键性质以及材料电子结构的重要工具。差分电荷、Bader电荷和Mulliken电荷是三种常用的电荷分析方法,它们分别从不同的角度描述了原子或分子中的电荷分布情况。以下将详细介绍这三种方法的定义、计算原理、优缺点以及应用场景。
差分电荷(Charge Density Difference, CDD)
定义: 差分电荷密度是通过比较体系总电荷密度与各独立组分电荷密度的差异,直观显示电子在相互作用中的重新分布。其数学表达式为: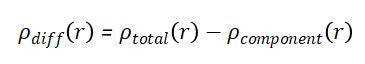 其中,ρtotal(r) 是整个体系的电荷密度,ρcomponent(r) 是各独立组分的电荷密度。差分电荷密度可以揭示电子在界面或吸附过程中的转移方向。
其中,ρtotal(r) 是整个体系的电荷密度,ρcomponent(r) 是各独立组分的电荷密度。差分电荷密度可以揭示电子在界面或吸附过程中的转移方向。物理意义:
黄色区域(电子积累):表明电子向该区域转移,通常出现在成键或吸附界面。
青色区域(电子耗散):表明电子从该区域流失,常见于原子核附近或断键区域。
计算方法:差分电荷密度通常通过第一性原理计算(如VASP、Quantum ESPRESSO等)获得。在计算过程中,需要分别计算吸附物和基底的电荷密度,然后进行差值计算。
应用场景:
吸附体系:如NO吸附在MgO/Mo表面,通过差分电荷分析发现电子从Mo基底向NO转移,表明化学键的形成。
界面反应:如H₂O在MgO/Mo表面的裂解,观察到电子在O–Mg界面的积累,驱动了水解离过程。
催化体系:如Co-Gr-Ca₂N和Fe-Gr-Y₂C体系中,差分电荷密度显示电介质基底向石墨烯层注入电子,增强了金属活性中心的电子密度。
优点:直观显示电子转移方向,便于理解界面相互作用。计算相对简单,适合快速分析。缺点:仅能提供定性信息,难以精确量化电荷转移量。在复杂几何结构中,电子积累和耗散区域可能难以区分。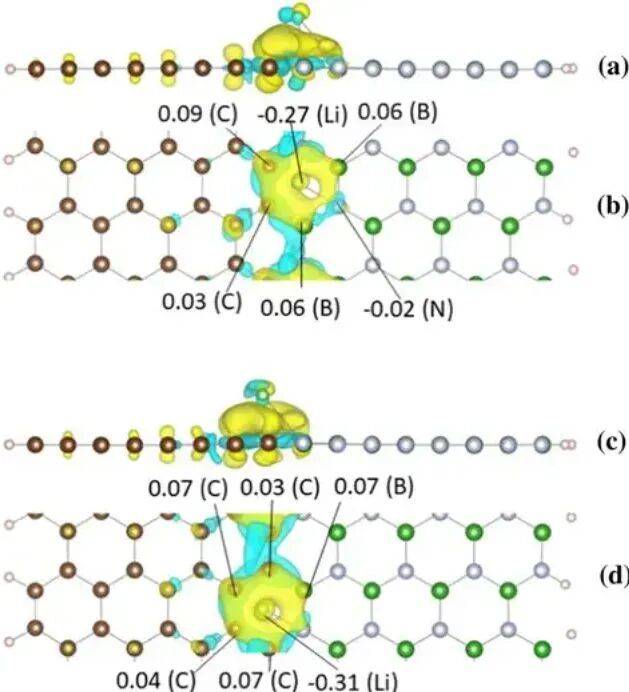
Bader电荷(Bader Charge)
定义:Bader电荷是基于AIM(Atoms in Molecules)理论,通过零通量面(zero-flux surface)划分原子体积,计算原子电荷。其公式为: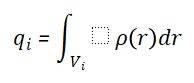 其中,Vi 是原子i的Bader体积,ρ(r) 是电荷密度。Bader电荷反映了原子在体系中的电荷状态,通常用于描述离子性材料或表面体系。
其中,Vi 是原子i的Bader体积,ρ(r) 是电荷密度。Bader电荷反映了原子在体系中的电荷状态,通常用于描述离子性材料或表面体系。计算方法:Bader电荷的计算需要先进行电子密度计算,然后通过AIM算法划分原子体积。常用的软件包括VASP、Multiwfn等。在计算过程中,需要设置LAECHG参数,并使用chgsum.pl和bader工具进行后处理。
物理意义:Bader电荷值反映了原子在体系中的电荷状态,通常与原子的化学环境密切相关。例如,在SiO₂中,Si的Bader电荷随配位数的增加而升高,从+2.0 e(二配位)到+3.18 e(四配位)。
应用场景:
离子性材料:如SiO₂、CaF₂等,Bader电荷可以准确描述离子键的特性。
表面研究:如Fe-Gr体系中,Bader电荷计算显示电介质基底向金属原子转移电子,优化了其d轨道电子占据,从而提高了催化活性。
电荷转移研究:如H₂O在MgO/Mo表面的裂解,Bader电荷计算显示O原子的电荷增加,表明其在反应中起关键作用。
优点:物理意义明确,适合周期性体系。能够定量分析电荷转移量,适用于催化、电池等领域的研究。
缺点:计算复杂,需要较高的计算资源。对共价键体系可能高估电荷,导致结果偏差。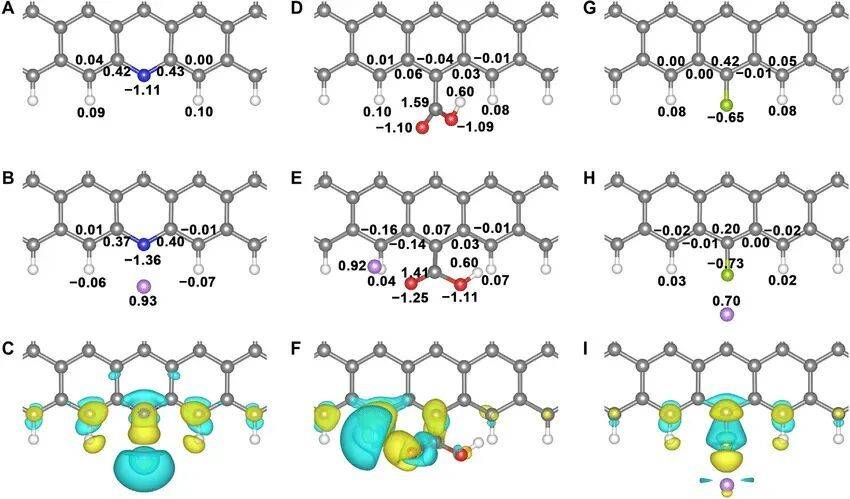
Mulliken电荷(Mulliken Charge)
定义:Mulliken电荷是基于分子轨道理论,通过将电子分配给各个原子,并计算电子占据数与核电荷之差来得到原子的电荷。其公式为: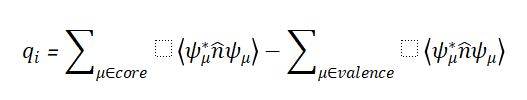 其中,ψμ 是原子轨道, 是电子数算符。Mulliken电荷通常通过量子化学软件(如Gaussian、ORCA等)计算。
其中,ψμ 是原子轨道, 是电子数算符。Mulliken电荷通常通过量子化学软件(如Gaussian、ORCA等)计算。计算方法:Mulliken电荷的计算基于分子轨道的展开,将电子分配给各个原子,并计算净原子布居和重叠布居。常用的基组包括6-31G、cc-pVDZ等,但基组的选择对结果影响较大。
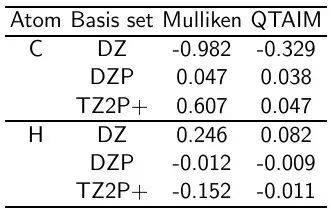
物理意义:Mulliken电荷值反映了原子在分子中的电荷状态,通常与原子的电负性相关。例如,在甲烷(CH₄)中,碳的Mulliken电荷随基组的增加而变化,从-0.329(DZ)到0.607(TZ2P+)。
应用场景:
分子体系:如TCNE分子中,Mulliken电荷显示中心原子的电荷较高,表明其在反应中起关键作用。
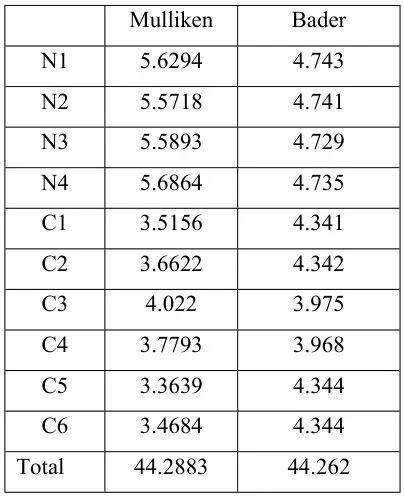
反应活性研究:如H₂O在MgO/Mo表面的裂解,Mulliken电荷计算显示O原子的电荷增加,表明其在反应中起关键作用。
电荷分布分析:如H₂O在MgO/Mo表面的裂解,Mulliken电荷计算显示O原子的电荷增加,表明其在反应中起关键作用。
优点:计算简单,适合快速分析。适用于分子体系,能够提供定性信息。
缺点:基组依赖性强,结果可能因基组选择而变化。物理意义不明确,难以定量分析。
三种电荷分析方法的比较