调控电势图以优化锂金属电池性能
调控电势图以优化锂金属电池性能 —— 基于电解液能量学的创新策略
引言:锂金属电池的潜力与挑战
在全球向绿色能源和碳中和技术转型的浪潮中,对高能量密度下一代储能设备的需求日益迫切。锂金属电池凭借锂金属极低的电极电势(-3.04 V vs 标准氢电极,SHE)和极高的理论容量(3860 mA・h・g⁻¹),成为最具潜力的高能量密度储能系统之一。然而,其广泛应用仍受限于循环稳定性不足的问题,而这一问题主要源于严重的电解液分解。
长期以来,科研人员致力于通过开发先进电解液(如高盐浓度电解液和局部高浓度电解液系统)解决这一难题。这些电解液通常包含耐还原醚类溶剂(如四氢呋喃(THF)、甘醇二甲醚)、功能性盐(如双氟磺酰亚胺锂(LiFSI))和稀释剂(如甲苯、氟化醚),其独特的离子对主导溶液结构赋予了它们诸多电化学优势。在传统的溶剂分离离子对(SSIP)电解液中,锂离子(Li⁺)被非质子溶剂溶剂化,与阴离子的相互作用较弱;而提高盐或稀释剂浓度会减少可用于 Li⁺溶剂化的游离溶剂分子,形成 Li⁺(阴离子)ₙ接触离子对(CIPs)和聚集体(AGGs),这一过程显著改变了电解液中所有物种间的库仑相互作用。
尽管这些研究取得了一定进展,但锂金属电池的可逆性仍面临重大挑战,这表明电解液设计中仍存在未解决的关键问题。本文基于 “调控电势图以优化锂金属电池” 的核心思路,系统阐述电解液能量学对电池性能的影响机制,揭示全电池电势平衡的重要性,并提出针对性的优化策略,为高稳定性、高能量密度锂金属电池的开发提供理论框架和实践指导。
电解液能量学:锂金属电池性能的核心调控因素
电解液中离子相互作用的双重影响
电解液中 Li⁺、阴离子与溶剂间的库仑相互作用是决定电池性能的核心因素,其通过两个关键途径影响锂金属电池的稳定性:
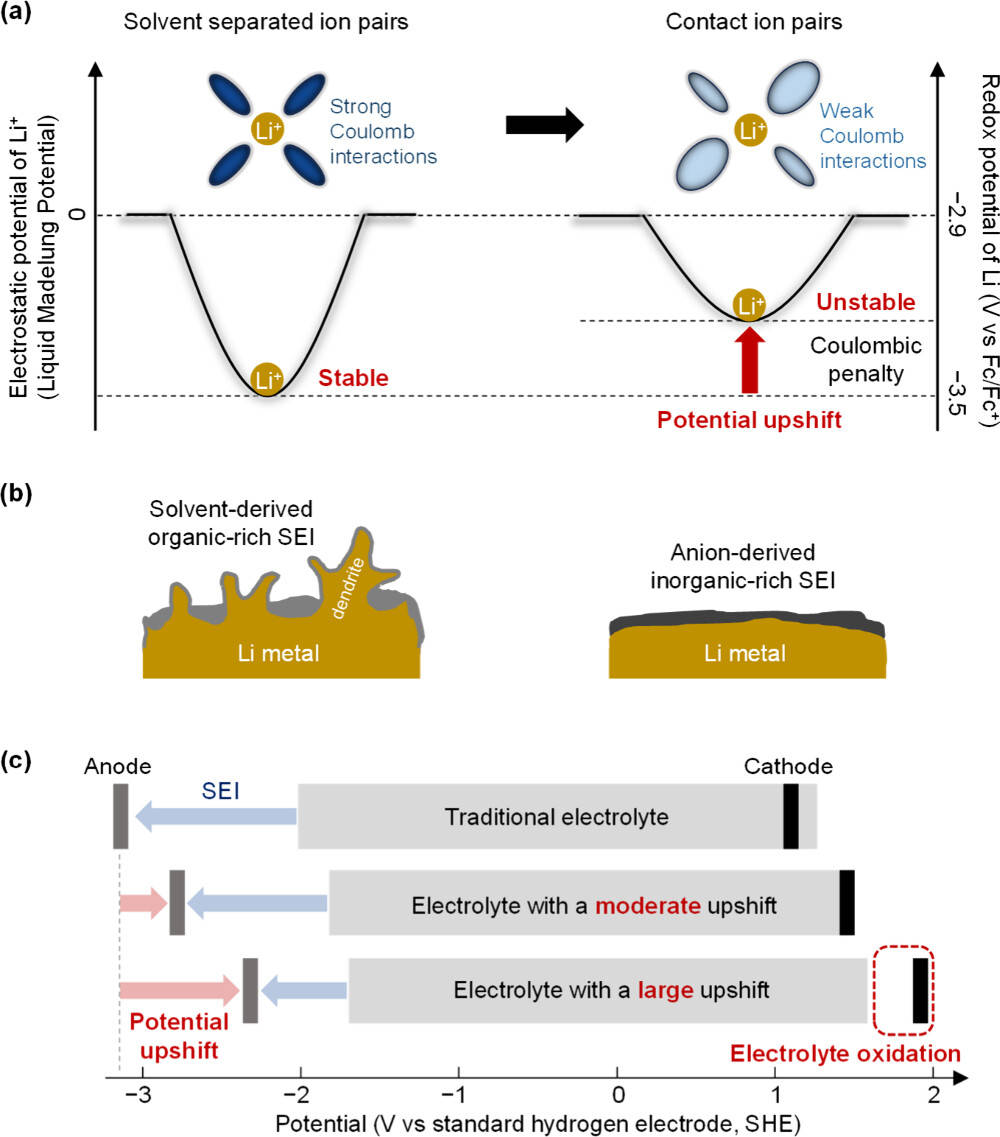
一方面,这种相互作用会改变 Li⁺的静电势能(即液体马德隆势),而液体马德隆势代表了 Li⁺与周围离子、溶剂和稀释剂之间的整体长程库仑相互作用。液体马德隆势的变化会导致锂金属的氧化还原电势(Eₗᵢ/ₗᵢ⁺)发生偏移,且这种偏移会以相同幅度同步影响阳极和阴极的反应电势。例如,当 Li⁺的配体从强相互作用物种(如强溶剂化溶剂)变为弱相互作用物种(如电子离域的大体积 FSI⁻阴离子或弱溶剂化溶剂 / 稀释剂)时,液体马德隆势会变得不稳定,进而导致 Eₗᵢ/ₗᵢ⁺向上偏移(电势上移)。
另一方面,离子相互作用会影响电解液的工作电势窗口。电势窗口的变化可能通过在电极表面形成钝化膜(如固体电解质界面相 SEI)引发动力学阻碍,而钝化膜的成分和稳定性直接决定了电解液与电极表面的反应程度。
基于定量实验结果系统平衡这两个因素,既能提升锂金属电池性能,又能最大限度减少电极表面的电解液降解 —— 这正是电解液能量学在电池设计中的核心价值。
离子对主导电解液对阳极可逆性的提升机制
在离子对主导的电解液中,离子相互作用的改变为提升锂金属阳极的可逆性带来了两个关键贡献:
Eₗᵢ/ₗᵢ⁺上移的热力学优势研究证实,在离子对主导的电解液中,锂金属的氧化还原电势(Eₗᵢ/ₗᵢ⁺)可上移超过 0.6 V,这一显著的电势偏移已通过实验研究、理论计算和机器学习得到验证。Eₗᵢ/ₗᵢ⁺上移能够降低电解液还原的热力学驱动力,减轻 SEI 的动力学支撑负担,从而在减少电解液分解的同时,实现锂沉积 / 剥离的高可逆性。
例如,在以 THF 为溶剂、LiFSI 为盐、甲苯为稀释剂的模型电解液中,随着稀释剂比例增加(THF: 甲苯的摩尔比从 10:0 调整为 3:7),拉曼光谱显示 CIP 和 AGG 的特征峰强度增加,而 SSIP 的特征峰强度降低,表明形成了富阴离子的溶剂化鞘。这种结构变化导致 Li⁺配体从电子定域物种向电子离域物种转变,通过破坏液体马德隆势引入库仑能量惩罚,最终使 Eₗᵢ/ₗᵢ⁺上移,提升了电解液的还原稳定性。
阴离子衍生 SEI 的动力学保护作用离子对主导的电解液中,Li⁺与阴离子的相互作用增强,会提高阳极表面附近的局部阴离子浓度,并降低阴离子的最低未占分子轨道(LUMO),从而加速阴离子优先于溶剂被还原。这种优先还原有助于形成机械和化学稳定性优异的阴离子衍生 SEI,阻止电解液与电极的直接接触,从动力学层面抑制锂金属表面的电解液还原。
X 射线光电子能谱(XPS)对循环后锂金属表面的分析结果证实了这一点:随着稀释剂用量增加,FSI⁻阴离子的还原碎片(如 Li-F、S-O-F、S-N-S 和 S-S)的信号更加显著。这种富无机成分的 SEI 能有效防止锂枝晶生长,进一步保障了锂沉积 / 剥离过程的稳定性。
综上,离子对主导的电解液通过 “Eₗᵢ/ₗᵢ⁺上移” 和 “阴离子衍生 SEI 形成” 的协同作用,从热力学和动力学两方面提升了锂金属阳极的稳定性,这是电解液设计的重要突破。
潜在风险:电势偏移引发的阴极降解问题
正极反应电势的同步偏移与氧化风险
尽管离子对主导的电解液能显著改善阳极性能,但研究发现,一种被忽视的电池降解模式可能在阴极发生 —— 这一问题由 Eₗᵢ/ₗᵢ⁺的大幅上移引发。
液体马德隆势的变化(如 Li⁺配体从强溶剂化溶剂向弱溶剂化稀释剂的转变)会导致所有与 Li⁺相关的反应电势以与 Eₗᵢ/ₗᵢ⁺相同的幅度偏移。这意味着,当 Eₗᵢ/ₗᵢ⁺上移时,阴极反应电势也会同步上移:虽然这会降低电解液还原的热力学驱动力,但会加速电解液氧化 —— 因为阴极反应电势可能超出电解液的工作电势窗口。
这种 “平行偏移” 在传统双电极电池系统中常被忽视,却可能导致阴极反应电势超出电解液的稳定范围,引发阴极表面的严重电解液降解。系统性实验(使用多种局部高浓度电解液评估电极电势偏移、电解液电势窗口与锂金属电池循环性能的关系)证实了这一被忽视的失效模式:Eₗᵢ/ₗᵢ⁺的大幅上移是阴极电解液氧化的关键诱因。
实验验证:不同电解液配比下的阴极稳定性差异
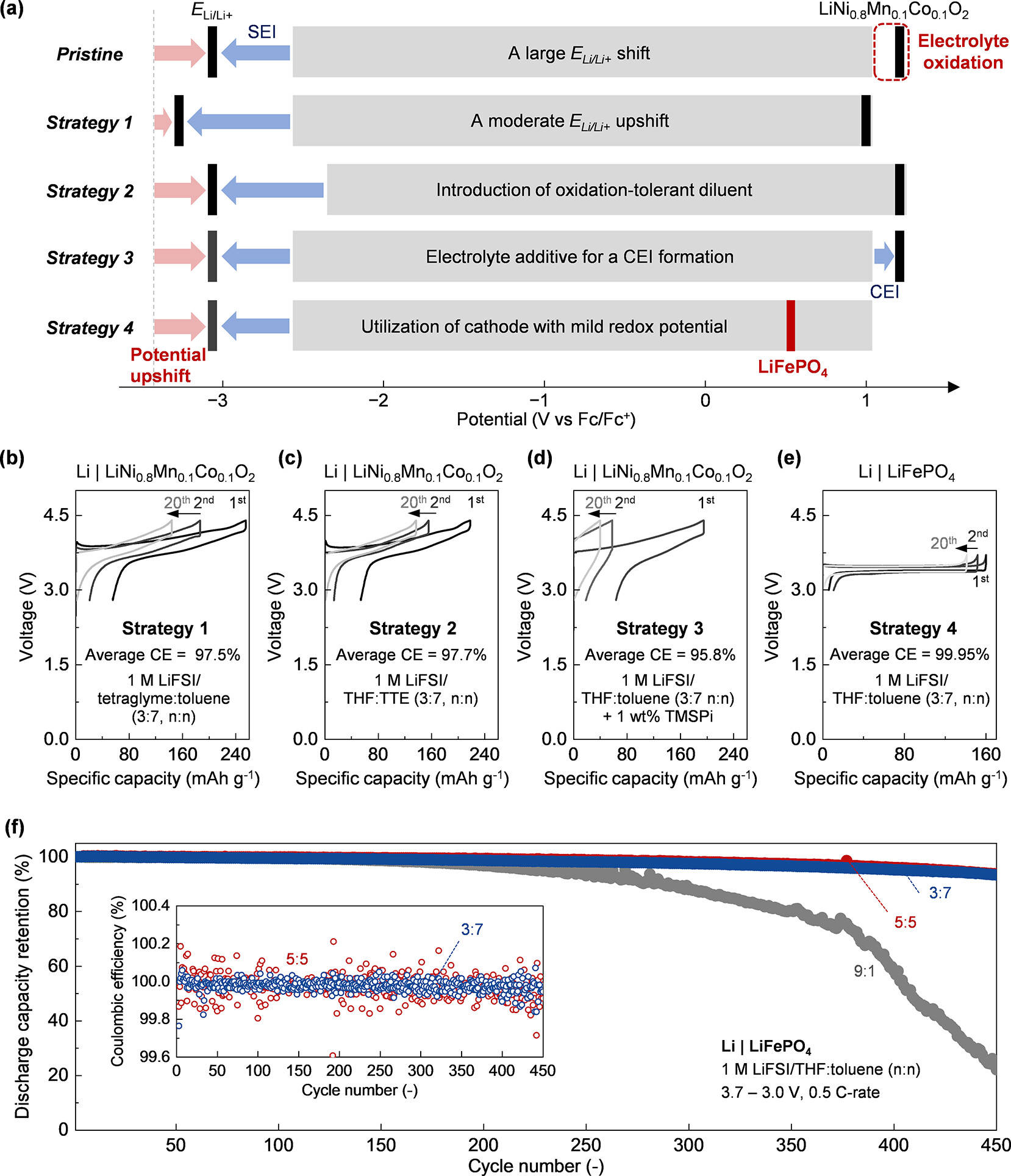
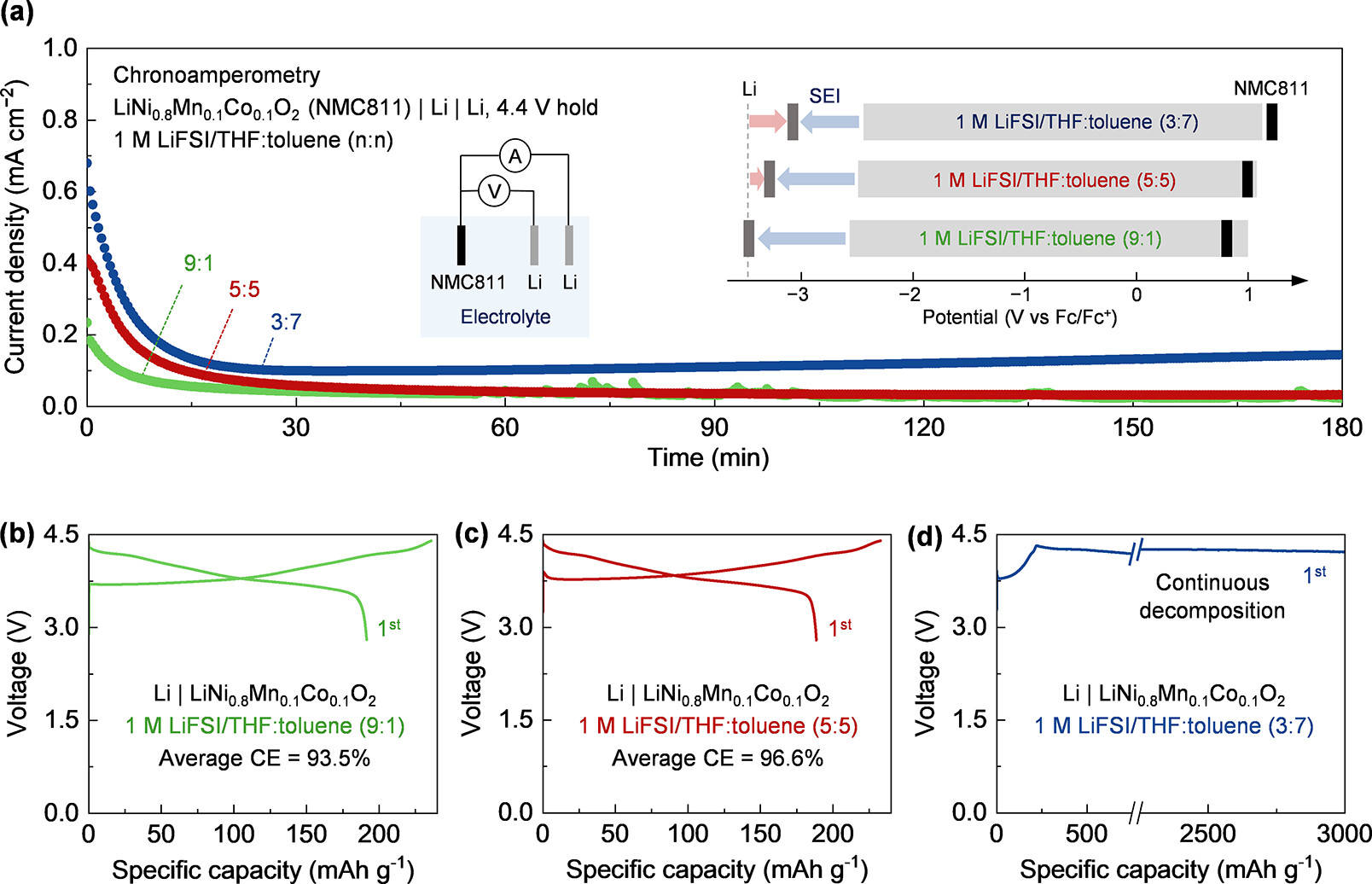
为验证上述机制,研究团队以 LiNi₀.₈Mn₀.₁Co₀.₁O₂(NMC811)为阴极、锂金属为阳极,在不同配比的 1 M LiFSI/THF: 甲苯电解液中进行了电化学测试:
电化学浮置(计时电流法,CA)测试在 4.4 V vs Li/Li⁺的条件下,1 M LiFSI/THF: 甲苯(3:7,n:n)电解液(Eₗᵢ/ₗᵢ⁺为 - 3.13 V vs Fc/Fc⁺,上移幅度较大)出现了显著更高的漏电流,这一现象归因于电解液氧化。相比之下,配比为 9:1 的电解液漏电流较小,表明阴极表面的氧化反应更弱。
恒流充放电测试在 Li|NMC811 全电池中,使用 1 M LiFSI/THF: 甲苯(3:7,n:n)电解液时,电池在 4.4 V 的上限截止电压下出现持续的电解液分解,无法实现可逆运行;而配比为 5:5 的电解液(Eₗᵢ/ₗᵢ⁺为 - 3.31 V vs Fc/Fc⁺,上移幅度适中)则表现出更高的库仑效率(96.6%)和更稳定的循环性能。
这些结果表明:大幅 Eₗᵢ/ₗᵢ⁺上移虽能稳定锂金属阳极,却会将 NMC811 阴极的反应电势推至电解液工作窗口之外,加速阴极表面的电解液氧化,最终导致电池失效。这一发现揭示了全电池设计中 “阳极 - 阴极平衡” 的重要性 —— 单纯优化阳极性能可能以牺牲阴极稳定性为代价。
优化策略:基于全电池电势图的电解液能量学调控
针对 “阳极稳定性提升与阴极氧化风险” 的矛盾,研究提出了四种基于全电池电势图的电解液能量学优化策略,通过系统性调控电势偏移和电解液窗口,实现正负极的协同稳定。
策略一:控制 Eₗᵢ/ₗᵢ⁺的适度上移
过度的 Eₗᵢ/ₗᵢ⁺上移会导致 THF 和甲苯在阴极表面氧化,解决这一问题的核心是通过调控 Li⁺与配体的库仑相互作用,加深液体马德隆势,实现 Eₗᵢ/ₗᵢ⁺的适度上移。
具体可通过两种方式实现:一是降低稀释剂的摩尔分数,增加系统中电子定域溶剂的浓度,从而诱导 Eₗᵢ/ₗᵢ⁺下移。实验显示,THF: 甲苯配比从 3:7 调整为 9:1 时,Eₗᵢ/ₗᵢ⁺从 - 3.13 V vs Fc/Fc⁺下移至 - 3.40 V vs Fc/Fc⁺,阴极氧化反应显著减弱。二是更换为具有更高溶剂化能力的溶剂,如甘醇二甲醚。四甘醇二甲醚含有多个电子定域氧原子,通过螯合效应与 Li⁺形成强配位 —— 在 1 M LiFSI / 四甘醇二甲醚:甲苯(3:7,n:n)电解液中,Eₗᵢ/ₗᵢ⁺仅为 - 3.41 V vs Fc/Fc⁺(上移幅度远小于 THF 体系),使 Li|NMC811 电池在 4.4 V 上限电压下实现可逆循环。
这种策略的核心是找到 “阳极稳定性” 与 “阴极耐受性” 的平衡点:适度的 Eₗᵢ/ₗᵢ⁺上移既能保留对阳极的保护作用,又能避免阴极反应电势超出电解液窗口。
策略二:引入耐氧化稀释剂
电解液的热力学氧化稳定性和还原稳定性存在权衡关系,单纯提升一方往往会牺牲另一方。引入耐氧化稀释剂为打破这一权衡提供了新思路 —— 以氟化醚(如 1,1,2,2 - 四氟乙基 - 2,2,3,3 - 四氟丙基醚,TTE)为例,其具有以下优势:
一是高氧化稳定性,可直接降低电解液在阴极表面的氧化程度;二是在充电初期会提前被还原,形成富氟 SEI,有助于抑制锂枝晶生长;三是能促进离子对形成,实现 Eₗᵢ/ₗᵢ⁺上移(1 M LiFSI/THF:TTE(3:7,n:n)中 Eₗᵢ/ₗᵢ⁺为 - 3.05 V vs Fc/Fc⁺,比 1 M LiFSI/THF: 甲苯(9:1,n:n)上移 350 mV)。
这种 “耐氧化 + 促离子对” 的双重特性,使 TTE 基电解液同时改善了锂金属阳极和高电势阴极的可逆性,为高电压锂金属电池提供了可行的电解液方案。
策略三:添加电解液添加剂构建阴极电解质界面相(CEI)
通过电解液添加剂在阴极表面形成功能性保护膜(CEI),可物理隔离阴极与电解液,减少直接接触引发的氧化反应。实验表明,在 1 M LiFSI/THF: 甲苯(3:7,n:n)电解液中添加 1 wt% 的三(三甲基硅基)亚磷酸酯(TMSPi)后,TMSPi 会优先在阴极表面分解,形成稳定的 CEI。
这种 CEI 不仅能阻止电解液与阴极的直接反应,还能屏蔽阴极活性材料免受氢氟酸(HF)的侵蚀 ——HF 是电解液水解的常见产物,会严重破坏阴极结构。需要注意的是,随着 Eₗᵢ/ₗᵢ⁺上移,CEI 的负担会增加(与 SEI 负担减少的趋势相反),因此添加剂的用量和种类需根据电势偏移幅度针对性调整。
策略四:匹配具有温和 redox 电势的阴极材料
商用电池需要在有限电解液用量下保证数千次循环的稳定性,而将石墨阳极替换为锂金属可使能量密度提升 50%~60%,这为选择温和电势阴极(如磷酸铁锂(LiFePO₄))提供了空间 —— 尽管其能量密度比 NMC811 低 20%~30,但整体系统能量密度仍能保持优势。
LiFePO₄的突出优势在于其温和的 redox 电势(3.4-3.5 V vs Li/Li⁺),这使其反应电势能稳定处于多数电解液的工作窗口内。实验显示,在 Li|LiFePO₄全电池中,使用 1 M LiFSI/THF: 甲苯(3:7,n:n)电解液(离子对更多,Eₗᵢ/ₗᵢ⁺上移更显著)时,电池在 450 次循环后容量保持率仍超过 85%,平均库仑效率达 99.95%;而使用 THF: 甲苯(9:1,n:n)电解液(离子对较少)时,容量保持率不足 20%,库仑效率约 99.59%。
这一结果证明:LiFePO₄的温和电势与离子对主导电解液的高 Eₗᵢ/ₗᵢ⁺形成了理想匹配 —— 既能充分发挥电解液对阳极的保护作用(高 Eₗᵢ/ₗᵢ⁺和阴离子衍生 SEI),又能避免阴极反应电势超出电解液窗口,是实现高稳定性锂金属电池的最实用方案之一。
实验验证:不同策略下的电池性能对比
为量化评估上述策略的有效性,研究团队通过库仑效率(CE)、循环容量保持率、电化学浮置测试等指标,对不同电解液体系和电池配置的性能进行了系统对比。
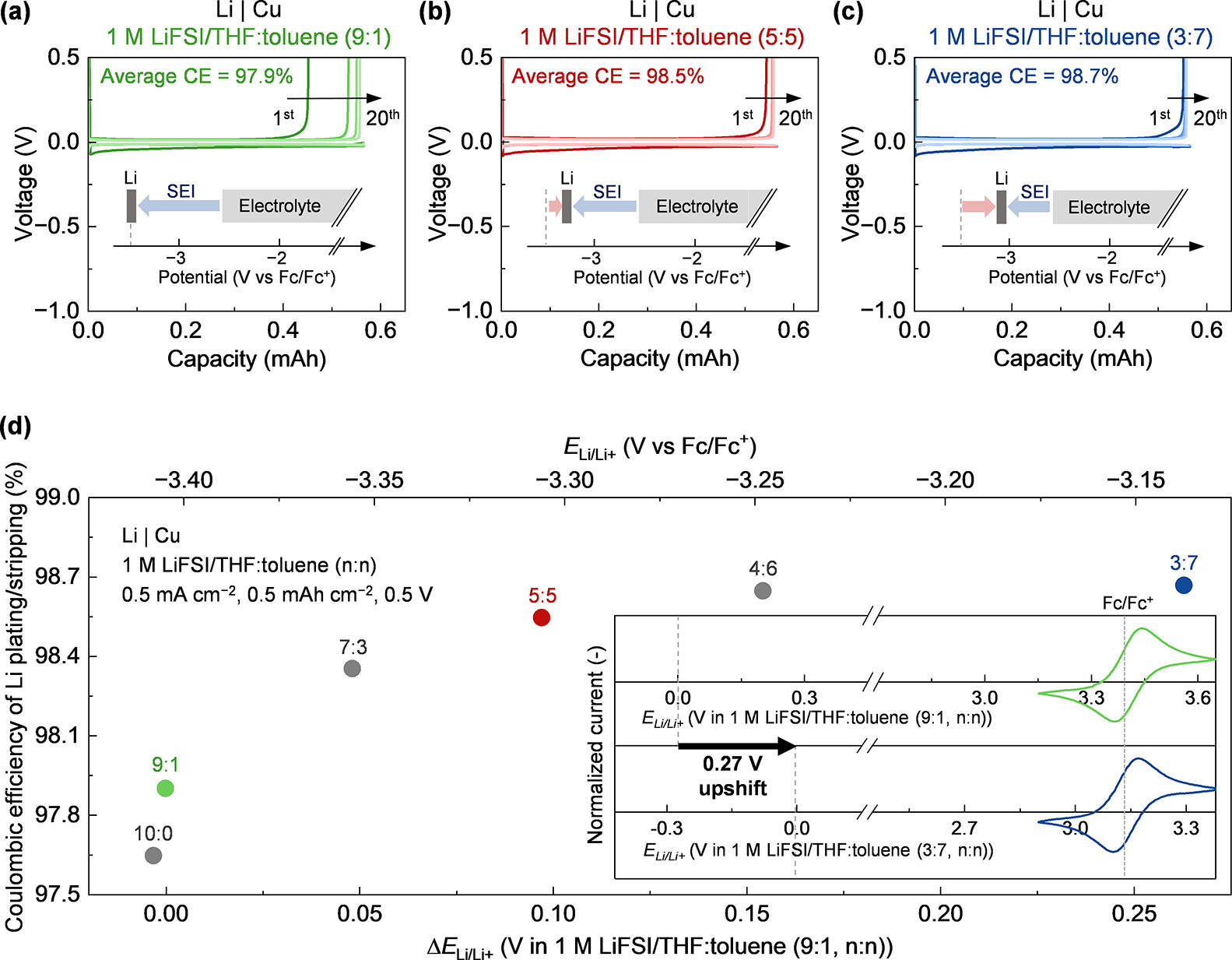
电解液配比对阳极性能的影响
在 Li|Cu 半电池中,随着 THF: 甲苯配比从 9:1 调整为 3:7(稀释剂增加),锂沉积 / 剥离的平均库仑效率逐步提升:9:1 配比时为 97.9%,5:5 配比时为 98.5%,3:7 配比时达到 98.7%。这一趋势与 Eₗᵢ/ₗᵢ⁺上移幅度一致 ——3:7 配比电解液的 Eₗᵢ/ₗᵢ⁺比 9:1 配比高 270 mV,证实了 Eₗᵢ/ₗᵢ⁺上移对阳极可逆性的提升作用。
值得注意的是,库仑效率计算中排除了第 1 次循环(因 SEI 形成消耗的不可逆锂和电子主要与界面亲和力、锂生长形貌等因素相关,与电解液热力学性质无关),更准确反映了电解液本身的稳定性。
全电池循环性能的策略差异
在 Li|NMC811 全电池中,不同策略的效果差异显著:
- 未优化的 3:7 配比甲苯电解液(大幅 Eₗᵢ/ₗᵢ⁺上移)因阴极氧化无法实现可逆循环;
- 采用四甘醇二甲醚替代 THF(策略一)后,电池平均库仑效率达 97.5%,循环稳定性显著提升;
- 使用 TTE 作为稀释剂(策略二)时,平均库仑效率进一步提升至 97.7%;
- 添加 TMSPi 添加剂(策略三)的电池平均库仑效率为 95.8%,虽低于前两者,但优于未优化体系。
而在 Li|LiFePO₄全电池中,3:7 配比甲苯电解液表现最佳:不仅容量保持率远高于其他配比,且库仑效率接近 100%,证明了 “高 Eₗᵢ/ₗᵢ⁺电解液 + 温和电势阴极” 组合的优越性。
表面分析与反应机制验证
XPS 对锂金属表面的分析显示,3:7 配比电解液中,锂表面的 Li-F、S-O 等无机物种信号更强,表明形成了更厚的阴离子衍生 SEI;而 9:1 配比电解液中有机物种(如 C-O)信号更显著,SEI 稳定性较差。
对 NMC811 阴极表面的分析则显示,3:7 配比甲苯电解液循环后的阴极表面存在大量 C=O 等氧化产物,证实了电解液氧化;而添加 TMSPi 或使用 TTE 稀释剂的阴极表面氧化产物显著减少,验证了 CEI 和耐氧化稀释剂的保护作用。
结论与展望
本研究揭示了一个隐藏但关键的电池降解机制:锂金属电池的性能提升需同时考虑全电池电势图和电解液能量学。离子对主导的电解液通过两种途径改善性能:一是破坏液体马德隆势使 Eₗᵢ/ₗᵢ⁺上移,二是促进阴离子衍生 SEI 形成,从热力学和动力学层面减少锂金属表面的电解液分解。
然而,Eₗᵢ/ₗᵢ⁺大幅上移会导致阴极反应电势超出电解液窗口,引发阴极氧化 —— 这一矛盾可通过四种策略解决:适度 Eₗᵢ/ₗᵢ⁺上移、使用耐氧化稀释剂、添加 CEI 形成添加剂,以及搭配 LiFePO₄等温和电势阴极。其中,LiFePO₄因其稳定的电势特性和成熟的应用基础,成为平衡正负极稳定性的最实用选择。
这些发现为电解液设计提供了新的指导思想:未来的锂金属电池优化不应局限于单一电极的性能提升,而需基于全电池电势平衡的理念,通过调控电解液能量学实现正负极的协同稳定。这一框架有望推动高能量密度锂金属电池的实用化,为新能源存储技术的发展奠定基础。
参考文献:
- Cheng, X.-B. et al. Toward Safe Lithium Metal Anode in Rechargeable Batteries: A Review. Chem. Rev. 2017, 117, 10403−10473.
- Zhang, X. et al. Towards Practical Lithium-Metal Anodes. Chem. Soc. Rev. 2020, 49, 3040−3071.
- Lin, D. et al. Reviving the Lithium Metal Anode for High-Energy Batteries. Nat. Nanotechnol. 2017, 12, 194−206.
- Borodin, O. et al. Uncharted Waters: Super-Concentrated Electrolytes. Joule 2020, 4, 69−100.
- Ko, S. et al. Electrode Potential Influences the Reversibility of Lithium-Metal Anodes. Nat. Energy 2022, 7, 1217−1224.
- Takenaka, N. et al. Liquid Madelung Energy Accounts for the Huge Potential Shift in Electrochemical Systems. Nat. Commun. 2024, 15, 1319.
- Park, G. et al. Strategy for Stable Interface in Lithium Metal Batteries: Free Solvent Derived vs Anion Derived. ACS Energy Lett. 2022, 7, 4274−4281.
- Padhi, A. K. et al. Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188−1194.